UnerfüllteR Kinderwunsch
Die Ursachen für einen unerfüllten Kinderwunsch sind vielfältig und können sowohl bei der Frau als auch beim Mann liegen. In einigen Fällen liegen genetische Veränderungen zugrunde, deren Nachweis es ermöglicht, gezielte therapeutische Maßnahmen zu ergreifen.
Untersuchung Anfordern
- Probenmaterial: EDTA-Vollblut (Molekulargenetik), Heparin-Vollblut (Chromosomenanalyse)
- Untersuchungsmethoden: Analyseabhängig (Allel-spezifische Genotypisierung, Fragmentlängenanalyse, MLPA, NGS)
- Benötigte Unterlagen:
Detaillierte Informationen zur Probeneinsendung finden sie hier.
Inhaltsverzeichnis
Allgemeine Informationen
Die Behandlung kinderloser Paare umfasst zahlreiche medizinische Aspekte sowohl auf Seiten der Frau als auch des Mannes. Die genetische Diagnostik bzw. die genaue Kenntnis der Ursache der Fertilitätsstörung stellt dabei einen wichtigen Baustein für die therapeutischen Optionen bei unerfülltem Kinderwunsch dar.
Die folgenden Informationen und Empfehlungen basieren auf den deutschen Leitlinien zur „Diagnostik und Therapie vor einer assistierten reproduktionsmedizinischen Behandlung“ (S2K-Level, AWMF Registry No. 015/085, 02/2019), die auch folgenden Algorithmus für die genetische Diagnostik bei Paaren mit unerfülltem Kinderwunsch empfehlen:
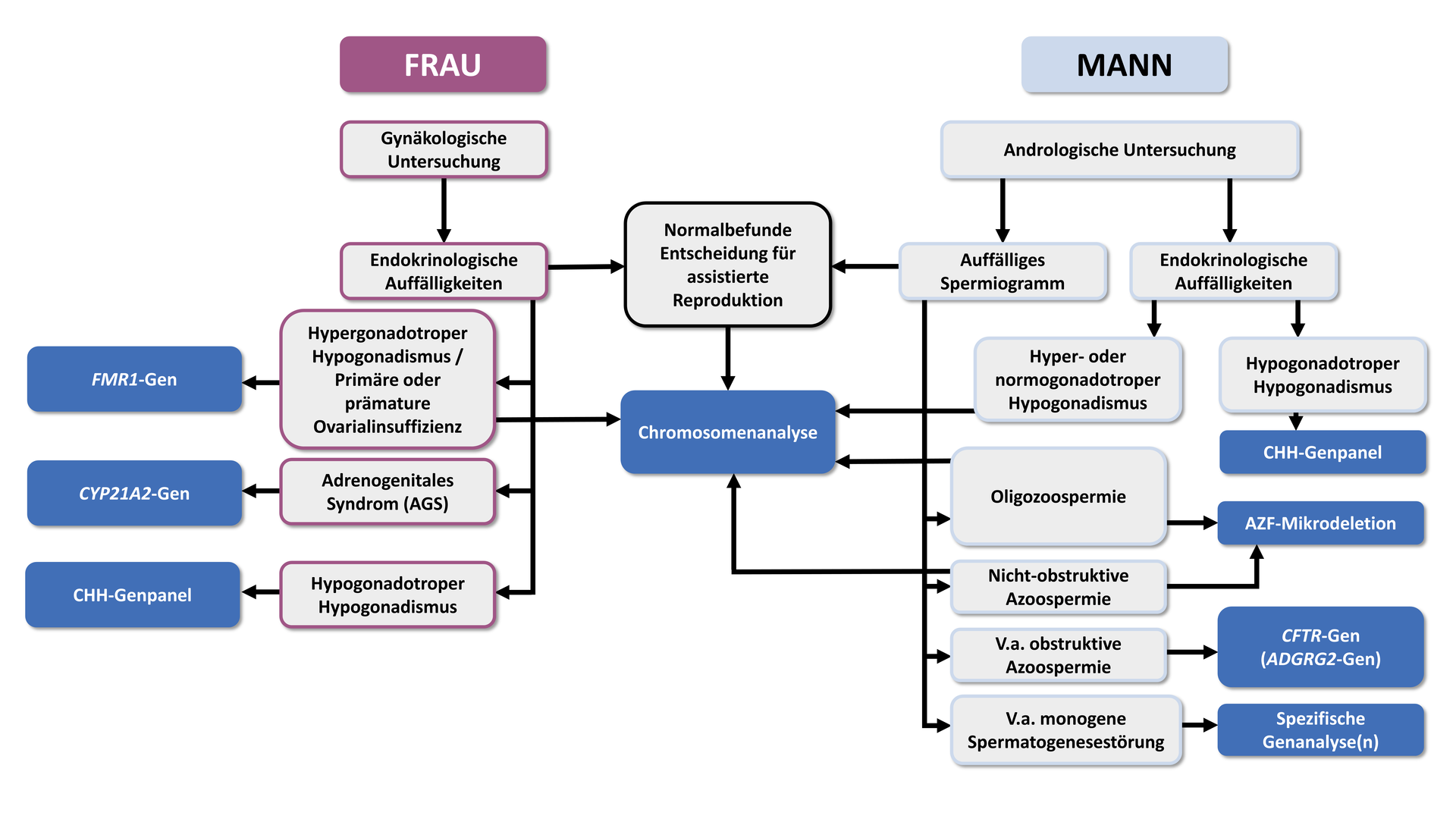
Genetische Ursachen beim Mann
Das wesentlichste Anzeichen für eine genetisch bedingte Ursache von Fruchtbarkeitsstörungen beim Mann ist ein pathologisches Spermiogramm. Bei Verdacht auf eine genetische Ursache, basierend auf den Ergebnissen einer grundlegenden andrologischen Diagnose, sollte eine spezifische genetische Untersuchung im Rahmen einer genetischen Beratung erwogen werden.
Spermatogenesesstörung
Spermatogenesestörungen sind Zustände oder Erkrankungen, die den normalen Prozess der Spermatogenese beeinträchtigen. Die Spermatogenese ist der biologische Prozess, bei dem sich Spermatogonien, die Stammzellen in den Hoden, durch verschiedene Entwicklungsstadien hindurch zu reifen Spermien entwickeln. Störungen in diesem Prozess können zu verschiedenen Problemen führen, die von einer verminderten Spermienproduktion (Oligozoospermie) bis hin zum vollständigen Fehlen von Spermien im Ejakulat (Azoospermie) reichen können.
Eine Azoospermie tritt dann auf, wenn entweder keine Spermien produziert werden (nicht obstruktive Azoospermie) oder sie aufgrund eines Hindernisses nicht in das Ejakulat gelangen können (obstruktive Azoospermie). Eine Oligozoospermie liegt hingegen vor, wenn die Spermienzahl im Ejakulat niedriger ist als normal (in der Regel weniger als 15 Millionen Spermien pro Milliliter Ejakulat).
Nicht obstruktive Spermatogenesestörungen
„Bei nicht-obstruktiver Azoospermie oder schwerer Oligozoospermie (<5 Mio/ml) soll nach Ausschluss anderer Ursachen eine Chromosomenanalyse erfolgen.“
Mikrodeletionen des Y-Chromosoms
Neben chromosomalen Aberrationen sind Mikrodeletionen in der Azoospermiefaktor-(AZF) Region des Y-Chromosoms eine der häufigsten genetischen Ursachen für eine nicht obstruktive Azoo- bzw. Oligozoospermie. Die Prävalenz von AZF-Deletionen liegt bei nicht-obstruktiver Azoospermie bei etwa 15-20% und etwa 7-10% bei schwerer Oligozoospermie. Innerhalb der AZF-Region befinden sich Gene (DAZ und RBM), die für die Spermatogenese von essentieller Bedeutung sind. Ein Funktionsverlust dieser Gene führt entweder zu einer Fehlentwicklung der Spermatogonien in den Hoden oder zur Entstehung unreifer, kondensierter Spermien.
Mikrodeletionen können in verschiedenen Regionen des AZF-Locus auftreten (AZFa, AZFb, AZFc). Eine präzise molekulargenetische Analyse und Beschreibung der Deletion ist von großer Bedeutung, insbesondere für Paare mit Kinderwunsch. Etwa 50% der Männer mit AZFc-Deletionen zeigen die Möglichkeit, durch eine testikuläre Spermienextraktion (TESE) Spermien zu gewinnen, im Gegensatz zu Patienten mit Deletionen in anderen Regionen, bei denen dies nicht der Fall ist.
„Bei nicht obstruktiver Azoospermie soll und bei schwerer Oligospermie (< 5 Mio/ml) sollte nach Ausschluss anderer Ursachen eine Analyse im Hinblick auf AZF-Mikrodeletionen (AZFa,b,c) erfolgen.“
Monogene Spermatogenesestörungen
In sehr seltenen Fällen können auch monogene Veränderungen ursächlich für eine nicht-obstuktive Azoospermie sein.
Bei Patienten ohne AZF-Deletion kann eine Analyse relevanter Gene als Teil einer Multi-Gen-Analyse mittels Next Generation Sequencing (NGS) in Betracht gezogen werden.
„Bei Verdacht auf eine seltene monogene Spermatogenesestörung kann eine genetische Analyse angeboten werden.“
Obstruktive Spermatogenesestörungen
Bei einer obstruktiven Azoospermie zeigt das Hodengewebe eine intakte Spermiogenese jedoch sind die Samenleiter blockiert oder nicht richtig ausgebildet. Diese kongenitale bilaterale Aplasie des Vas deferens (CBAVD) oder seltener eine kongenitale unilaterale Aplasie des Vas deferens (CUAVD) liegt bei ca. 2% aller Männer mit Azoospermie vor. In den meisten Fällen sind dafür Mutationen im CFTR-Gen (ursächlich für Mukoviszidose) verantwortlich. Bei etwa 80% der Männer mit einer CBAVD kann mindestens eine Mutation im CFTR-Gen nachgewiesen werden. Da nachweislich nur homozygote oder compound-heterozygote Mutationen zu einer CBAVD führen, sollte in diesen Fällen eine Komplettsequenzierung des CFTR-Gens (einschließlich 5-T Allel) erfolgen. Dies ist auch für die Risikoabschätzung für zukünftige Kinder entscheidend. Liegt eine CFTR-assoziierte CBAVD vor, sind die Erfolgsaussichten für assistierte Reproduktionstechniken wie TESE oder ICSI vielversprechend.
Mit dem ADGRG2-Gen ist ein weiterer Locus bekannt, der ursächlich für eine CBAVD sein kann.
„Bei Verdacht auf eine obstruktive Azoospermie soll nach Ausschluss anderer Ursachen eine Analyse des CFTR-Gens erfolgen. Diese soll alle relevanten pathogenen Mutationen incl. des TG-T-Repeats in Intron 8 erfassen; falls damit nur eine heterozygote Mutation gefunden wird, soll eine vollständige Sequenzierung erfolgen.“*
„Sofern bei einer obstruktiven Azoospermie die CFTR-Analyse einen unauffälligen Befund erbracht hat, sollte eine Analyse des ADGRG2-Gens erfolgen.“
Genetische Ursachen bei der Frau
Etwa 40% der Frauen, die an Unfruchtbarkeit leiden, sind von einer ovulatorische Dysfunktion betroffen. In diesen Fällen ist eine gründliche Untersuchung der hormonellen Achsen indiziert, um das Vorliegen eines hypo- oder hypergonadotroper Hypogonadismus sowie einer Hyperandrogenämie zu analysieren.
Hypergonadotroper Hypogonadismus
Hypergonadotroper Hypogonadismus bei Frauen kann aufgrund verschiedener genetischer Ursachen auftreten, die die normale Funktion der Eierstöcke beeinträchtigen können. Bei etwa 10% der Frauen liegt eine Gonosomenaberration (häufig auch im Mosaik) wie das Turner Syndrom (45,X) oder das Triple-X-Syndrom (47,XXX) vor.
„Bei Frauen mit hypergonadotroper Hypogonadismus soll nach Ausschluss anderer Ursachen eine Chromosomenanalyse durchgeführt werden.“
Ovarialinsuffizienz
Prämutationen im FMR1-Gen, die durch CGG-Repeats mit einer Länge von 55–200 charakterisiert sind, sind mit einem erhöhten Risiko für primäre oder sekundäre Ovarialinsuffizienz assoziiert. Wenn eine solche Prämutation an die Nachkommen weitergegeben wird, besteht außerdem eine hohe Wahrscheinlichkeit, dass sich bei Kindern eine Vollmutation entwickelt (Repeatlängen > 200). Vollmutationen im FMR1-Gen sind ursächlich für das Fragile-X-Syndrom, das mit leichter bis schwere Intelligenzminderung einhergeht und mit Verhaltensstörungen und charakteristischen körperlichen Merkmalen verbunden sein kann.
Bei Frauen mit primärer Ovarialinsuffizienz, im speziellen mit auffälliger Familienanamnese, liegt bei bis zu 15% eine FMR1-Prämutation vor.
„Bei primärer oder prämaturer Ovarialinsuffizienz soll nach Ausschluss anderer Ursachen eine genetische Analyse der CGG-Wiederholungen im FMR1-Gen durchgeführt werden.“
Hypogonadotroper Hypogonadismus
Hypogonadotroper Hypogonadismus (CHH) bei Frauen, ist durch einen Mangel an gonadotropen Hormonen gekennzeichnet (insbesondere an follikelstimulierendes Hormon FSH und luteinisierendes Hormon LH), die normalerweise von der Hypophyse im Gehirn produziert werden. Diese Hormone spielen eine entscheidende Rolle bei der Regulation der Eierstockfunktion und der Produktion von Östrogen und Progesteron.
Die Prävalenz von kongenitalem hypogonadotropem Hypogonadismus bei Frauen liegt zwischen 1:30.000 und 1:40.000. Mittlerweile können bis zu 40% der molekularen Ursachen dieses Zustands auf Mutationen in etwa 20 Genen zurückgeführt werden, darunter GNRHR, FSHB, LEP/LEPR, LHB, FGFR1.
„Bei Frauen mit kongenitalem hypogonadotropem Hypogonadismus (CHH) kann nach Ausschluss exogener Ursachen eine genetische Analyse der CHH-Gene durchgeführt werden.“
Hyperandrogenämie
Hyperandrogenämie bezeichnet einen erhöhten Spiegel männlicher Sexualhormone im Blut, wobei das Adrenogenitale Syndrom (AGS) die vorherrschende genetische Ursache dafür ist. Die am häufigsten vorkommende Form ist der autosomal rezessive 21-Hydroxylase-Mangel, der durch Mutationen im CYP21A2-Gen verursacht wird. Eine genetische Testung ist zu empfehlen, da sich die endokrine Betreuung nach dem zugrundeliegenden Enzymdefekt bzw. der Art der nachgewiesenen Mutationen richtet.
„Bei Verdacht auf ein adrenogenitales Syndrom soll eine genetische Diagnostik durchgeführt werden.“
Genetische Ursachen beim Paar
KIR- und HLA-C Genotypen
Die Interaktion zwischen natürlichen Killerzellen (NK-Zellen) in der Gebärmutterschleimhaut und dem sich entwickelnden Embryo gehört zu den immunologischen Faktoren, welche das Zustandekommen und den Fortbestand einer Schwangerschaft wesentlich beeinflussen. Vermittelt wird diese Interaktion durch Killerzell-Immunoglobulin-ähnliche Rezeptoren (KIR), die von uterinen NK-Zellen exprimiert werden, und HLA-C-Antigenen auf der Oberfläche des Trophoblasten. Sowohl die KIR-Genfamilie als auch die HLA-C-Moleküle weisen dabei eine äußerst hohe genetische Variabilität auf. Obwohl das komplexe Zusammenspiel der beiden Gensysteme noch nicht umfassend geklärt ist, gibt es Hinweise darauf, dass bestimmte Kombinationen von mütterlichen KIR-Genotypen und fetalen HLA-C-Molekülen ein höheres Risiko für Implantationsversagen, habituelle Aborte und Schwangerschaftskomplikationen wie Präeklampsie mit sich bringen.
Die KIR- beziehungsweise HLA-C-Genotypisierung im Rahmen einer reproduktionsmedizinischen Behandlung erfolgt durch eine Analyse der genomischen DNA des Kinderwunschpaares. Dabei wird untersucht, welche Sets an aktivierenden und inhibierenden KIR bei der Frau vorhanden sind, und der HLA-C-Genotyp des Mannes (HLA-C1/C1, HLA-C1/C2 oder HLA-C2/C2) bestimmt. Sollte für die Behandlung eine Gametenspende erforderlich sein, wird auch die Eizellspenderin beziehungsweise der Samenspender einer HLA-C-Genotypisierung unterzogen.
Das immunologische Verständnis der KIR-HLA-C-Interaktion reicht aktuell nicht aus, um eine kausale Beziehung zwischen bestimmten elterlichen Genotypen und dem Auftreten von Implantationsversagen, wiederholten Fehlgeburten oder Schwangerschaftskomplikationen zweifelsfrei herzustellen. Verschiedene Studien zeigen jedoch, dass die aus der KIR- und HLA-Genotypisierung gewonnenen Informationen dabei helfen können
- die optimale Anzahl an Embryonen für einen Transfer im IVF-Zyklus zu bestimmen
- möglichen Komplikationen während der Schwangerschaft vorzubeugen und
- im Fall einer Gametenspende eine Spenderin beziehungsweise einen Spender auszuwählen, deren/dessen HLA-C-Genotyp mit dem KIR-Genotyp der zukünftigen Mutter immunologisch kompatibel ist