Infertility
The causes of infertility are diverse and may lie with either the woman or the man. In some cases, underlying genetic changes can be identified, enabling targeted therapeutic measures.
Request Test
- Sample material: EDTA whole blood (molecular genetics), heparin whole blood (chromosome analysis)
- Test methods: Depending on the analysis (allele-specific genotyping, fragment length analysis, MLPA, NGS)
- Required documents:
Detailed information on sample submission can be found here.
Table of contents
General information
The treatment of childless couples involves numerous medical aspects on both the female and male side. Genetic diagnostics, or precise knowledge of the cause of the fertility disorder, is an important component of the therapeutic options for infertility.
The following information and recommendations are based on the German guidelines on “Diagnostics and therapy prior to assisted reproductive medical treatment” (S2K level, AWMF Registry No. 015/085, 02/2019), which also recommend the following algorithm for genetic diagnostics in couples with infertility:
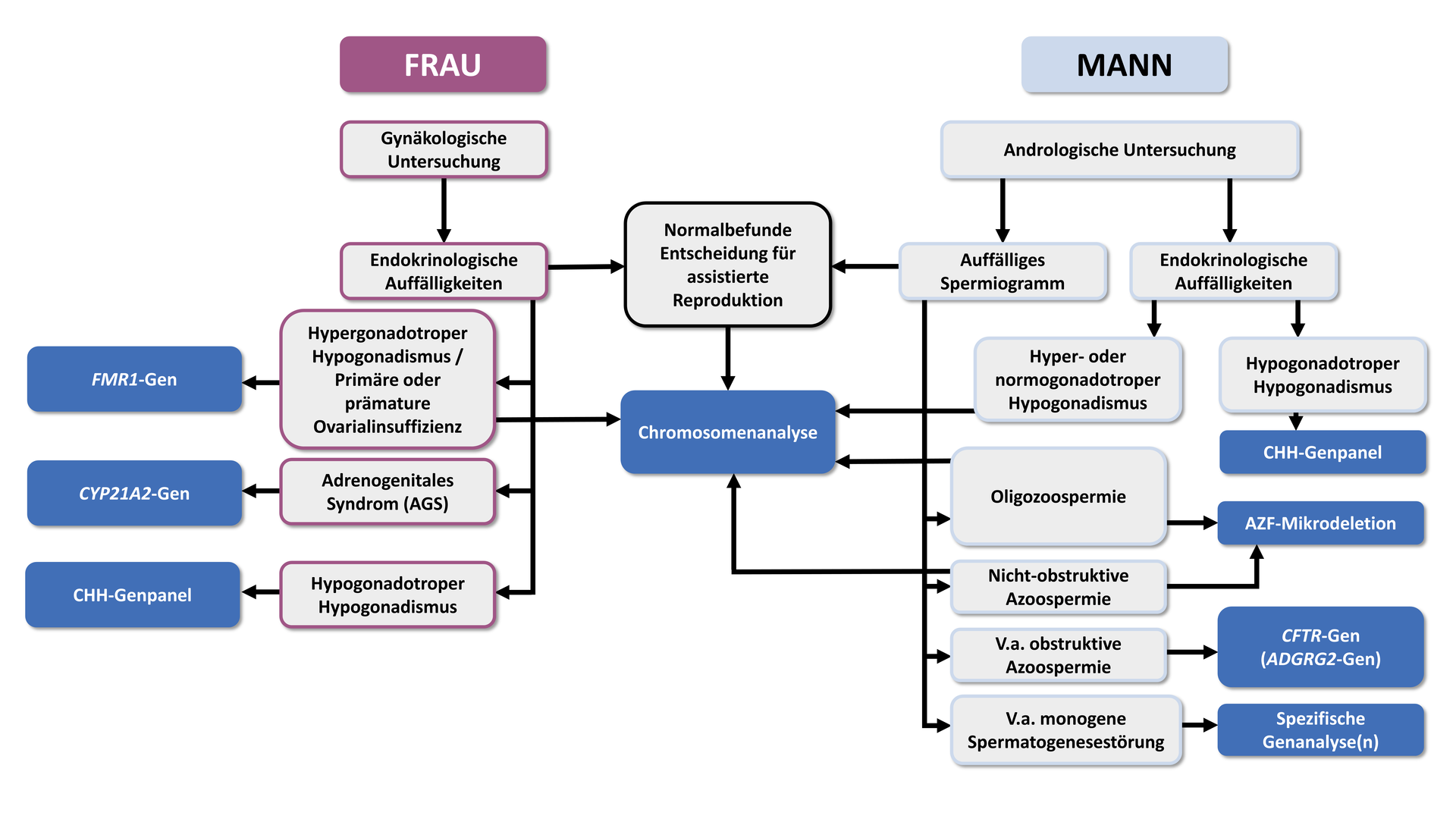
Genetic causes in men
The most important indicator of a genetically caused fertility disorder in men is an abnormal semen analysis. If a genetic cause is suspected based on the results of a basic andrological assessment, a specific genetic test should be considered as part of genetic counselling.
Disorders of spermatogenesis
Disorders of spermatogenesis are conditions or diseases that impair the normal process of spermatogenesis. Spermatogenesis is the biological process in which spermatogonia, the stem cells in the testes, develop into mature sperm through various developmental stages. Disruptions in this process can lead to a range of problems, from reduced sperm production (oligozoospermia) to the complete absence of sperm in the ejaculate (azoospermia).
Azoospermia occurs when either no sperm are produced (non-obstructive azoospermia) or they cannot reach the ejaculate due to an obstruction (obstructive azoospermia). Oligozoospermia, by contrast, is present when the sperm count in the ejaculate is lower than normal (usually fewer than 15 million sperm per millilitre of ejaculate).
Non-obstructive disorders of spermatogenesis
“In non-obstructive azoospermia or severe oligozoospermia (<5 million/ml), chromosome analysis should be performed after other causes have been excluded."
Y-chromosome microdeletions
In addition to chromosomal aberrations, microdeletions in the azoospermia factor (AZF) region of the Y chromosome are among the most common genetic causes of non-obstructive azoo- or oligozoospermia. The prevalence of AZF deletions is approximately 15–20% in non-obstructive azoospermia and about 7–10% in severe oligozoospermia. The AZF region contains genes (DAZ and RBM) that are essential for spermatogenesis. Loss of function of these genes leads either to abnormal development of spermatogonia in the testes or to the formation of immature, condensed sperm.
Microdeletions can occur in different regions of the AZF locus (AZFa, AZFb, AZFc). A precise molecular genetic analysis and description of the deletion is of great importance, particularly for couples wishing to conceive. About 50% of men with AZFc deletions may be able to obtain sperm via testicular sperm extraction (TESE), in contrast to patients with deletions in other regions, where this is not the case.
“In non-obstructive azoospermia, and in severe oligospermia (< 5 million/ml), after other causes have been excluded, an analysis for AZF microdeletions (AZFa,b,c) should be performed.”
Monogenic disorders of spermatogenesis
In very rare cases, monogenic variants may also be the cause of non-obstructive azoospermia.
In patients without an AZF deletion, analysis of relevant genes as part of a multi-gene panel using next-generation sequencing (NGS) may be considered.
“If a rare monogenic disorder of spermatogenesis is suspected, genetic analysis may be offered.”
Obstructive disorders of spermatogenesis
In obstructive azoospermia, testicular tissue shows intact spermiogenesis, but the vas deferens are blocked or not properly developed. This congenital bilateral absence of the vas deferens (CBAVD), or more rarely congenital unilateral absence of the vas deferens (CUAVD), is present in about 2% of all men with azoospermia. In most cases, mutations in the CFTR gene (causative for cystic fibrosis) are responsible. In around 80% of men with CBAVD, at least one mutation in the CFTR gene can be detected. Since only homozygous or compound heterozygous mutations have been shown to lead to CBAVD, complete sequencing of the CFTR gene (including the 5T allele) should be performed in these cases. This is also crucial for risk assessment for future children. If CFTR-associated CBAVD is present, the prospects of success for assisted reproductive techniques such as TESE or ICSI are promising.
The ADGRG2 gene is another locus known to be causative for CBAVD.
“If obstructive azoospermia is suspected, analysis of the CFTR gene should be performed after other causes have been excluded. This should include all relevant pathogenic mutations, including the TG-T repeat in intron 8; if only a heterozygous mutation is found, complete sequencing should be performed.”*
“If CFTR analysis in obstructive azoospermia yields an unremarkable result, analysis of the ADGRG2 gene should be performed.”
Genetic causes in women
Approximately 40% of women who experience infertility are affected by ovulatory dysfunction. In these cases, a thorough evaluation of the hormonal axes is indicated to assess the presence of hypo- or hypergonadotropic hypogonadism as well as hyperandrogenaemia.
Hypergonadotropic hypogonadism
Hypergonadotropic hypogonadism in women can occur due to various genetic causes that may impair normal ovarian function. In about 10% of women, a sex chromosome aberration (often also mosaic) such as Turner syndrome (45,X) or triple X syndrome (47,XXX) is present.
“In women with hypergonadotropic hypogonadism, chromosome analysis should be performed after other causes have been excluded.”
Ovarian insufficiency
Premutations in the FMR1 gene, characterised by CGG repeats with a length of 55–200, are associated with an increased risk of primary or secondary ovarian insufficiency. If such a premutation is passed on to offspring, there is also a high likelihood that a full mutation will develop in children (repeat lengths > 200). Full mutations in the FMR1 gene are causative for fragile X syndrome, which is associated with mild to severe intellectual disability and may be accompanied by behavioural disorders and characteristic physical features.
In women with primary ovarian insufficiency, especially those with a notable family history, an FMR1 premutation is present in up to 15%.
“In primary or premature ovarian insufficiency, genetic analysis of CGG repeats in the FMR1 gene should be performed after other causes have been excluded.”
Hypogonadotropic hypogonadism
Congenital hypogonadotropic hypogonadism (CHH) in women is characterised by a deficiency of gonadotropic hormones (in particular follicle-stimulating hormone, FSH, and luteinising hormone, LH), which are normally produced by the pituitary gland in the brain. These hormones play a crucial role in regulating ovarian function and the production of oestrogen and progesterone.
The prevalence of congenital hypogonadotropic hypogonadism in women is between 1:30,000 and 1:40,000. Up to 40% of the molecular causes of this condition can now be attributed to mutations in around 20 genes, including GNRHR, FSHB, LEP/LEPR, LHB, and FGFR1.
“In women with congenital hypogonadotropic hypogonadism (CHH), genetic analysis of CHH genes may be performed after exogenous causes have been excluded.”
Hyperandrogenaemia
Hyperandrogenaemia refers to an increased level of male sex hormones in the blood, with congenital adrenal hyperplasia (CAH) being the predominant genetic cause. The most common form is autosomal recessive 21-hydroxylase deficiency caused by mutations in the CYP21A2 gene. Genetic testing is recommended, as endocrine management depends on the underlying enzyme defect and the type of mutations detected.
“If congenital adrenal hyperplasia is suspected, genetic diagnostics should be performed.”
Genetic causes in the couple
KIR and HLA-C genotypes
The interaction between natural killer cells (NK cells) in the uterine lining and the developing embryo is among the immunological factors that significantly influence the establishment and maintenance of pregnancy. This interaction is mediated by killer cell immunoglobulin-like receptors (KIR) expressed by uterine NK cells and HLA-C antigens on the surface of the trophoblast. Both the KIR gene family and HLA-C molecules exhibit extremely high genetic variability. Although the complex interplay between the two gene systems has not yet been fully elucidated, there is evidence that certain combinations of maternal KIR genotypes and fetal HLA-C molecules are associated with a higher risk of implantation failure, recurrent miscarriage, and pregnancy complications such as pre-eclampsia.
KIR and HLA-C genotyping as part of reproductive medical treatment is performed by analysing the genomic DNA of the couple wishing to conceive. This assesses which sets of activating and inhibitory KIR are present in the woman, and determines the man’s HLA-C genotype (HLA-C1/C1, HLA-C1/C2, or HLA-C2/C2). If gamete donation is required for treatment, the egg donor or sperm donor will also undergo HLA-C genotyping.
Current immunological understanding of the KIR–HLA-C interaction is not sufficient to conclusively establish a causal relationship between specific parental genotypes and the occurrence of implantation failure, recurrent miscarriages, or pregnancy complications. However, various studies show that the information obtained from KIR and HLA genotyping can help to
- determine the optimal number of embryos for transfer in the IVF cycle
- help prevent possible complications during pregnancy, and
- in the case of gamete donation, select a donor whose HLA-C genotype is immunologically compatible with the future mother’s KIR genotype